
MPS stanowi wielkie wyzwanie dla medycyny. Chorzy na mukopolisacharydozę na pierwszy rzut oka wyglądają jak osoby niskorosłe. To jednak poważna, wielonarządowa choroba o ciężkim przebiegu, która poważnie zagraża życiu! Jak rozpoznać MPS i jak ją leczyć?
[aktualizacja: 07’2020]
Mukopolisacharydoza – rzadka, genetyczna choroba o ciężkim przebiegu! Co wiemy o MPS?
Mukopolisacharydoza (MPS) to choroba wielonarządowa trudna do diagnozy i leczenia. Osobę chorą na to rzadkie schorzenie na pierwszy rzut oka ocenić, jako niskorosłą. MPS jest jednak chorobą, która atakuje wiele narządów stwarzając poważne zagrożenie dla życia! Niestety ta przypadłość dotyka zarówno dzieci, jak i dorosłych i nie ma prostego wyjaśnienia podłoża jej występowania.
Mukopolisacharydoza (MPS) to choroba metaboliczna, w której dochodzi do nadmiernego gromadzenia w różnych tkankach związków zwanych mukopolisacharydami.
Kluczową rolę w przypadku MPS odgrywa wnikliwa obserwacja pacjenta i rzetelnie przeprowadzony wywiad lekarski, który może nasunąć właściwe rozpoznanie! To podstawa do dalszego postępowania diagnostycznego i holistycznego podejścia do leczenia mukopolisacharydozy.
Za przyczynę występowania MPS uznaje się wadę metabolizmu, na skutek której w organizmie dochodzi do kumulowania się mukopolisacharydów uszkadzających komórki i narządy.
W przebiegu mukopolisacharydozy organizm nie jest w stanie wytwarzać wystarczającej ilości enzymów odpowiedzialnych za rozkładanie mukopolisacharydów. Dotychczas zidentyfikowano 11 różnych niedoborów enzymatycznych, które powodują siedem odrębnych MPS. W wyniku niedoboru enzymu w lizosomach wielu komórek organizmu gromadzą się związki chemiczne GAG (glikozaminoglikany).
Na skutek gromadzenia się glikozaminoglikanów dochodzi do uszkodzeń narządów.
W efekcie dochodzi do zaburzeń funkcjonowania komórek, co może powodować uszkodzenia narządów takich jak: serce, kości, stawy, układ oddechowy i układ nerwowy. Nieprawidłowości mogą być widoczne od razu po urodzeniu, jednak ich objawy rozwijają się powoli na skutek postępujących uszkodzeń! Skutki kumulacji GAG mogą być różne w różnych MPS. W wielu przypadkach występują cechy kliniczne o różnym stopniu nasilenia. Warto wiedzieć, że każde zaburzenie może obejmować szeroki wachlarz nasilenia klinicznego. MPS może przyjmować postać wolno postępującą (co nie oznacza łagodniejszego przebiegu) lub postać charakteryzującą się szybszą progresją.
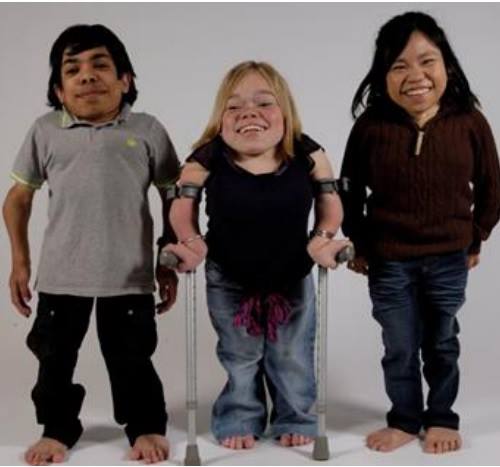
Mukopolisacharydoza – choroba wieloukładowa o szeregu pozornie nie związanych ze sobą objawów!
U tego samego pacjenta mogą współwystępować objawy kliniczne w różnym stopniu ciężkości. Zwykle są to: choroby serca, niedobór wzrostu, zmętnienie rogówki, zaburzenia widzenia, sztywność i bóle stawów, głuchota, częste zakażenia ucha oraz znaczące zajęcie układu kostnego (ręka szponiasta, dysostosis multiplex). Ponadto MPS objawia się również: zespołem zaburzeń kostnych, przepukliną pępkową i pachwinową, uciskiem promieniowo-szczytowo-potylicznym oraz niższą przewidywalną długością życia.
Zaburzenia kostne, takie jak szablowata kość łokciowa i skolioza, widoczne w badaniach RTG, stanowią wspólny mianownik wszystkich postaci MPS.
Mukopolisacharydoza charakteryzuje się również zaburzeniami wieloukładowymi:
- sztywność stawów (dla wszystkich typów MPS za wyjątkiem MPS IV, gdzie występuje wiotkość stawów),
- choroba zastawkowa serca,
- niskorosłość,
- niestabilność kręgosłupa szyjnego,
- ucisk rdzenia kręgowego,
- choroby układu oddechowego,
- utrata słuchu,
- zmętnienie rogówki,
- nieprawidłowości stomatologiczne,
- upośledzona wytrzymałość fizyczna.
Warto zwrócić uwagę, że we większości przypadków mukopolisacharydozy upośledzenie intelektualne nie dotyczy MPS IV i VI. Osoby te często cechuje wręcz błyskotliwa inteligencja pozwalająca osiągać sukcesy edukacyjne i zawodowe.
Jednym z najważniejszych aspektów MPS jest fakt, że choroba wiąże się z różnorodnymi fenotypami.
Oznacza to, że ta sama choroba może przybierać różne postacie. U niektórych pacjentów nasilone objawy występują już od najwcześniejszych lat życia, u innych natomiast objawy postępują powoli. U pacjentów z wolno postępującymi postaciami choroby występują zmiany tkankach o zróżnicowanym nasileniu, w związku z czym mukopolisacharydoza może być trudna w rozpoznaniu. Oznacza to także mniejsze możliwości leczenia i mniejsze szanse na poprawę stanu pacjenta, zwłaszcza w przypadku objawów kardiologicznych! Z tego powodu zawsze konieczna jest całościowa ocena pacjenta z uwzględnieniem wszystkich aspektów mukopolisacharydozy. W szczególności istotne są: jakość życia, funkcjonowanie kończyn oraz związana z tym mobilność i wytrzymałość, a także zdolność do samodzielnego funkcjonowania.
Diagnostyka mukopolisacharydozy
Diagnozowanie mukopolisacharydozy odbywa się w oparciu o wyniki kliniczne w połączeniu z wynikami badań laboratoryjnych. Diagnozę można potwierdzić wyłącznie za pośrednictwem odpowiednich badań laboratoryjnych, takich jak analiza aktywności enzymu. Złotym standardem są testy aktywności enzymatycznej, wymagane w ramach programów lekowych. W niektórych programach oprócz testu enzymatycznego wymagany jest również test molekularny.
Rozpoznanie mukopolisacharydozy wymaga wykonania odpowiednich badań klinicznych i laboratoryjnych.
Wstępną i najprostszą metodą diagnostyczną jest ilościowe oznaczenie mukopolisacharydów w moczu oraz określenie ich rodzaju. Zastosowanie mają różne metody ilościowe (np. test bibułowy, test zmętnieniowy) i jakościowe (np. elektroforezę). Ilość mukopolisacharydów w moczu (zarówno osób chorych, jak i zdrowych) zależy od wieku pacjenta, stężenia moczu oraz wielu innych czynników. Niekiedy badanie należy powtórzyć, aby mogło być w pełni miarodajne.
Dodatkową metodą stosowaną w niektórych przypadkach mukopolisacharydozy, np. do określenia nosicielstwa, jest identyfikacja mutacji, czyli analiza DNA izolowanego np. z krwi pacjenta.
W zależności od tego, który enzym nie działa lub działa źle, gromadzą się różne mukopolisacharydy, co prowadzi do rozróżnienia kilku typów mukopolisacharydoz określanych jako:
- Zespół Hurlera (MPS IH), Hurlera-Scheiego (MPS IH/S) i Scheiego (MPS IS)
- Choroba Hunter (MPS II)
- Syndrom Sanfilippo (MPS III)
- Choroba Morquio (MPS IV)
- Zespół Maroteaux-Lamy’ego (MPS VI)
- Zespół Sly’a (MPS VII)
- Choroba Natowicza (MPS IX) – obecnie uznawana za najrzadszy ze wszystkich typów MPS
Choroby rzadkie na świecie i w Polsce. Możliwości leczenia mukopolisacharydozy.
Obecnie znanych jest na świecie ponad 6 000 różnych chorób rzadkich, które dotykają sumarycznie od 6% do 8% populacji. Populacja Polska wynosi 38,2 mln osób, z czego 2,3 – 3 mln jest dotknięta chorobami rzadkimi. Dla porównania ok. 2 mln osób choruje w Polsce na cukrzycę.
Z medycznego punktu widzenia dostrzegalne są jedynie te choroby rzadkie, które są leczone przy użyciu leków, a to stanowi tylko 5% populacji dla pozostałych, czyli dla 95% nie ma terapii farmakologicznej!
Z legislacyjnego punktu widzenia choroby rzadkie są niezauważone, ponieważ brakuje jakichkolwiek ustawowych regulacji uwzględniających ich specyfikę. Polska jest krajem, w którym odnotowuje się najniższy poziom refundacji leków sierocych w UE, wykorzystywanych w leczeniu chorób rzadkich.
Leki sieroce (orphan medicinal products) to leki desygnowane przez COMP (Committee for Orphan Medicinal Products) stosowane w terapiach rzadkich. Dzięki tej desygnacji tego typu leki mają inne wymagania rejestracyjne wynikające z rzadkości występowania choroby, liczebności populacji, przebiegu itd.
W Unii Europejskiej zarejestrowanych jest 116 produktów leczniczych dedykowanych chorobom rzadkim. W Polsce refundacji podlega tylko 14 z nich. To najniższy poziom refundacji leków sierocych w krajach UE!
Pacjenci z chorobami rzadkimi, takimi jak mukopolisacharydoza (MPS) nie mają równego dostępu do leczenia. Tylko 3 typy MPS I, MPS II i MPS VI objęte są refundacją w ramach programów lekowych!
Specyfika chorób rzadkich powinna być uwzględniana w wymaganiach wobec analizy HTA i procesie ich oceniania przez AOTMiT. Powinno być brane pod uwagę dopuszczenie innych niż dotychczasowe dowodów naukowych (badania nierandomizowane, case studies, ocena efektu klinicznego na podstawie surogatów itp.)
Dostępność do specjalisty i jakość prowadzenia pacjenta z mukopolisacharydozą
Wieloukładowość mukopolisacharydozy jest wyzwaniem zarówno dla pacjenta, jaki i dla lekarza zarówno na etapie diagnozy oraz w trakcie procesu kontrolowania choroby. Należy podkreślić fakt, iż w Polsce brakuje specjalistów zajmujących się dorosłymi pacjentami z chorobami metabolicznymi. Leczenie MPS głównie polega na leczeniu objawowym, mającym za zadanie złagodzenie symptomów.
Nie ma skutecznych terapii hamujących lub cofających chorobę – obecnie całkowite wyleczenie mukopolisacharydozy nie jest możliwe (nie istnieje tzw. terapia przyczynowa)
Łagodzenie objawów niektórych chorób współistniejących (nie pełne wyleczenie!) można osiągnąć poprzez przeszczep szpiku kostnego. To zabieg bardzo dużego ryzyka, jednak ma zastosowanie w MPS I i u niektórych pacjentów z MPS II (choroba Huntera). W mukopolisacharydozach typu III (Sanfilippo) i typu IV (Morquio) zabieg jest nieskuteczny.
Inną metodą jest tzw. zastępcza terapia enzymatyczna, polegająca na bezpośrednim dożylnym podawaniu oczyszczonego enzymu. Metodami inżynierii genetycznej możliwe jest produkowanie na skalę techniczną i odpowiednia modyfikacja enzymów brakujących w niektórych chorobach lizosomalnych. Od kilku lat zastępczą terapię enzymatyczną stosuje się w przypadkach MPS I, MPS II, MPS VI, obecnie MPS IV – tylko MPS I, MPS II i MPS VI są objęte w Polsce refundacją. Pacjenci z MPS IV czekają na refundację.
Mukopolisacharydoza i co dalej? Rokowania pacjentów z MPS
Niestety rokowania pacjentów z MPS nie są optymistyczne. Głównymi czynnikami odpowiadającymi za wysoką śmiertelność są: powikłania płucne, niestabilność kręgosłupa szyjnego i mielopatia szyjna, powikłania kardiologiczne, powikłania mające związek z operacją chirurgiczną i znieczuleniem ogólnym, w przypadku którego utrudnieniem jest wąska i kręta tchawica oraz zbyt obfita wydzielina.
Obecnie trwają prace nad przygotowaniem Narodowego Planu Chorób Rzadkich przez powołany w Ministerstwie Zdrowia Zespół pod przewodnictwem wiceministra zdrowia Krzysztofa Łandy. Narodowy Plan Chorób Rzadkich ma pozwolić na zapewnienie trwałej realizacji polityki zdrowotnej ukierunkowanej na potrzeby pacjentów z chorobami rzadkimi, wprowadzić systemowe rozwiązania problemów zdrowotnych i socjalnych tej grupy chorych.
Wierzymy, że powołani do zespołu eksperci podejdą do tematu rzetelnie i z dużą dozą empatii, aby wypracować plan w pełni uwzględniający potrzeby pacjentów z chorobami rzadkimi. Każdy Polak ma bowiem zagwarantowane prawo do ochrony zdrowia oraz do pomocy w razie choroby lub niezdolności do pracy!
Materiał powstał we współpracy ze Stowarzyszeniem Chorych na Mukopolisacharydozę (MPS) i Choroby Rzadkie.
Więcej o chorobach rzadkich, metodach diagnostycznych oraz sposobach ich leczenia przeczytasz w naszym dziale: Choroby rzadkie.
Podcast do wysłuchania
źródło: mat. prasowe GREEN COMMUNICATION. Materiał powstał we współpracy ze Stowarzyszeniem Chorych na Mukopolisacharydozę (MPS) i Choroby Rzadkie.
Naszym gościem był dr hab. n. med. Zbigniew Żuber, specjalista pediatra i reumatolog, pracujący w Wojewódzkim Specjalistycznym Szpitalu Dziecięcym im. św. Ludwika w Krakowie





